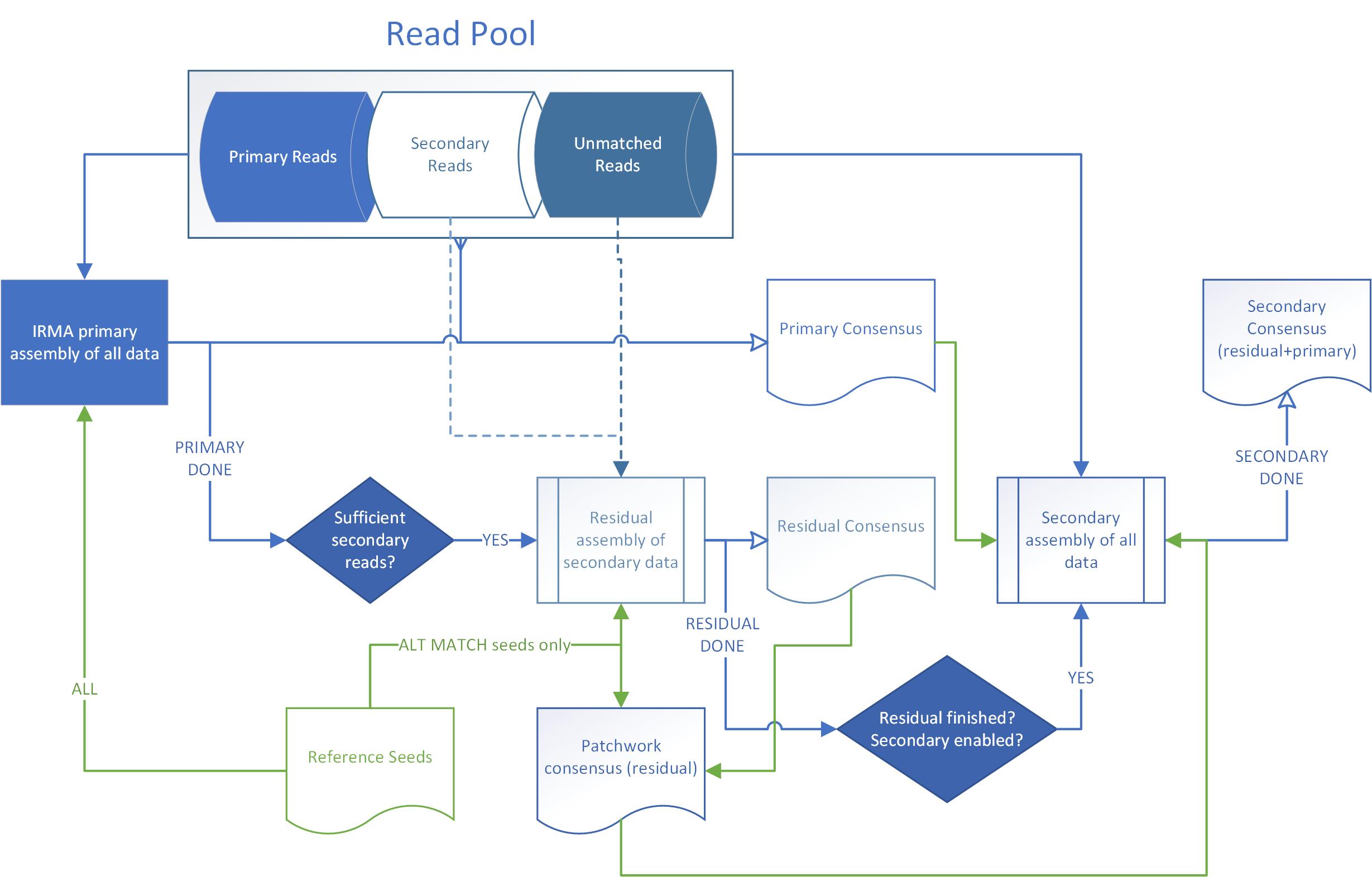
Residual and secondary assembly
A residual assembly is an assembly that builds consensus strictly from secondary and unmatched data after the primary assembly has finished. Data must be separable as specified by the SORT_GROUPS option, with reference seeds available as usual. For example, suppose A_HA_H3 and A_HA_H5 are both present in the FLU module and that SORT_GROUPS uses "HA" as a pattern for gene name sorting. If a sample contained reads for both gene segments, then A_HA_H3 competes with A_HA_H5 to be in the primary assembly. The HA with the most reads will be primary, say A_HA_H3 in this case. Secondary data will be stored for A_HA_H5. Three options control if and how a residual assembly is triggered: RESIDUAL_ASSEMBLY_FACTOR, MIN_RC_RESIDUAL, and MIN_RP_RESIDUAL.
Read pattern counts (de-duplicated reads) for all primary data are compared to all read patterns from secondary data. If there are enough read patterns in the secondary data, IRMA tries to do the residual assembly. First, an observed factor is calculated, which is the number of primary read patterns divided by secondary ones. As the factor gets large, the secondary data gets small. For example, suppose A_HA_H3 (primary) had 2000 read patterns and A_HA_H5 (secondary) had 200 read patterns (and only HA is detected in this sample). Then the observed factor as an integer is 10 (2000 / 200). This is another way of saying secondary data is 10% of the size of primary data. If the observed factor is less than the RESIDUAL_ASSEMBLY_FACTOR, residual assembly is tried. By default, RESIDUAL_ASSEMBLY_FACTOR is set to 0, preventing residual assembly from being tried. In our influenza module we have a configuration option that sets RESIDUAL_ASSEMBLY_FACTOR equal to 400, so the observed factor must be <400 (or >0.25% secondary data compared to primary) in order to trigger a residual assembly. Spurious residual data is to be expected due to extremely low-level contamination and misclassification. The dividing line between trace levels and significant signal is up to the user.
During the first round of residual assembly, each gene segment requires a minimum number of reads counts and patterns to be matched for the assembly to continue. This is necessary because the observed factor could be artificially low due to low overall read count in the sample. Moreover, only one of the secondary data's genes may have sufficient reads for assembly. The minimum read count and pattern thresholds, MIN_RC_RESIDUAL, and MIN_RP_RESIDUAL respectively, control this check. We recommend higher thresholds than the primary assembly MIN_RC and MIN_RP defaults to avoid spurious residual assemblies. Our default configuration sets these to 150 for the residual assembly threshold versus 15 in the primary assembly.
Residual assembly output structure mimics the output structure for primary assembly, but will appear as subfolder residual_assembly within the main output folder.
Secondary assembly performs a brand new assembly on the same sample given both primary and residual assembly data exist and DO_SECONDARY is set to 1. In a secondary assembly, reads are restored to the original state and the reference seeds are set to the consensus sequences from both the primary and residual final output. These consensus should be from disjoint sets of genes. For residual data, it is possible some of the reads missing may be due to partial homology with the primary dataset, leading to missing loci. If profile HMMs are provided for the same genes, IRMA uses them to align residual and reference seed consensus and build a "patch-work consensus". In particular, the patch-work consensus algorithm only uses the aligned reference seed sequence for filling in the "holes" in the residual assembly consensus.
Aside from the change in reference seeds, primary and secondary meta-assembly run a bit differently from one another in terms of the match and sort steps. First, in the match step the BLAT_IDENTITY variable is raised from 80% to 96%. Since IRMA now gathers reads matching previous final consensus, we expect them to be quite close to reference. We also want to disallow homologous genes from picking up spurious reads that could mislead the sort step. Secondly, the sort step automatically sets SORT_GROUPS="", meaning all genes with non-negligible reads are assembled as primary rather than sifting some into a secondary data designation. Homologous genes, particularly if reads contain mutations, will compete with one another for assembly data. Highly accurate sorting (e.g., SORT_PROG="LABEL") is therefore encouraged but not enforced.
Secondary assembly output structure mimics the output structure for primary assembly, but will appear as subfolder secondary_assembly within the main output folder.
Return to the Influenza Division Bioinformatics Team homepage