Understanding IRMA output
Output for IRMA is deposited into the working directory of the calling user as a new subfolder. This subfolder is named using the sample name argument. If the subfolder already exists with the same name, IRMA will add a suffix in the format of "-V#", incrementing the number until it finds a folder that doesn't exist. Below is typical output from a sample in our manuscript, but we have excluded all gene segments except A_MP for brevity. Hover over underlined text for an explanation of the output file or folder; click on links under the figures/ and tables/ folder to see output samples and explanations.
Return to the IRMA homepage
IRMA output directory structure (only showing A_MP)
Mixture_Example/Sample name
│
├── amended_consensus/Assembled consensuses
per gene segment w/mixed base calls.
│ └── Mixture_Example_7.faAmended
consensus.
│ └── Mixture_Example_7.a2mOptional amended
global alignment to profile HMM.
†
│ └── Mixture_Example_7.pad.faOptional
N-padded consensus for amplicon dropouts.
†
│
├── figures/
│ ├── A_MP-coverageDiagram.pdf
Shows coverage and variant calls.
│ ├── A_MP-heuristics.pdfHeuristic graphs for A_MP
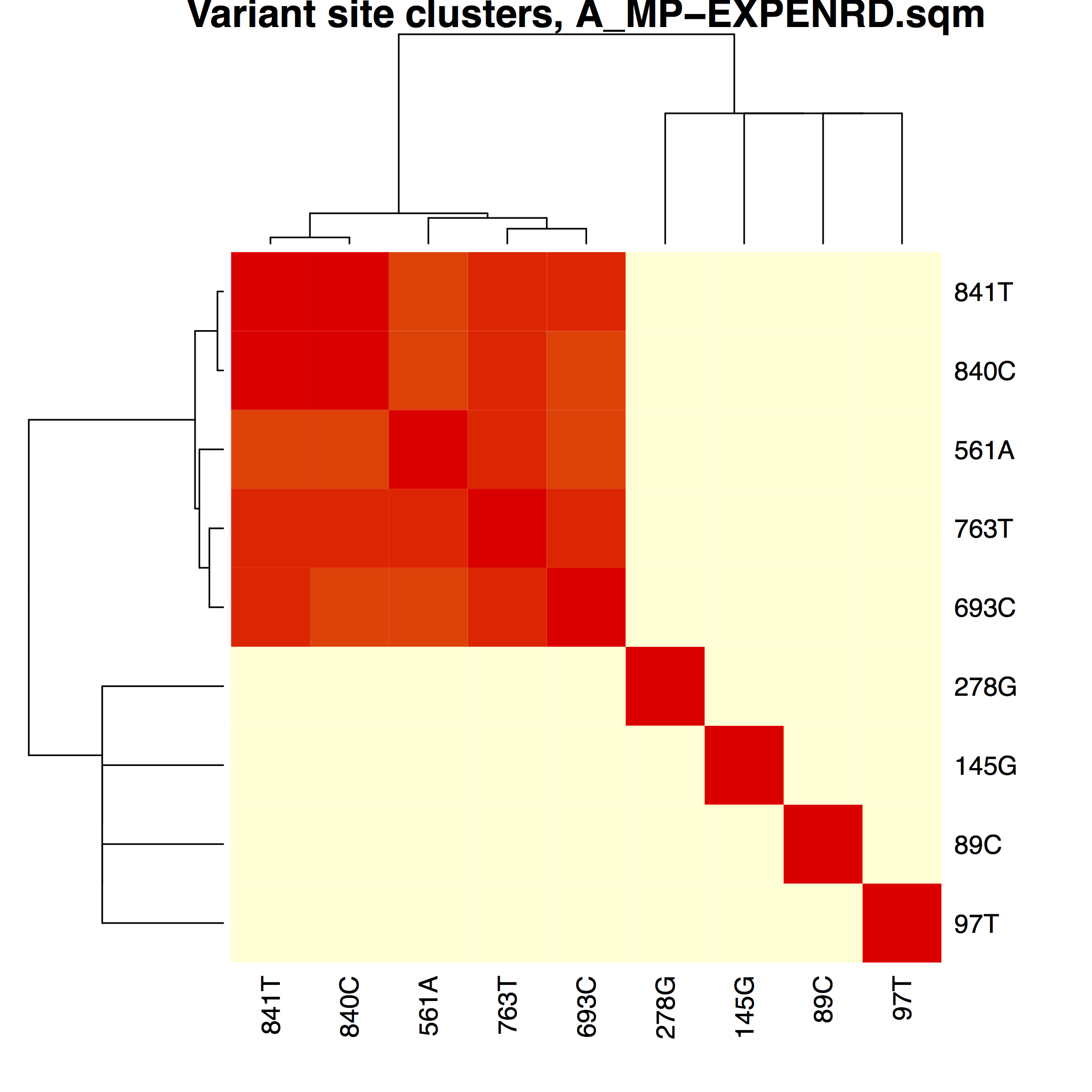
│ ├── A_MP-EXPENRD.pdfA_MP variant phasing using 'experimental enrichment" distances
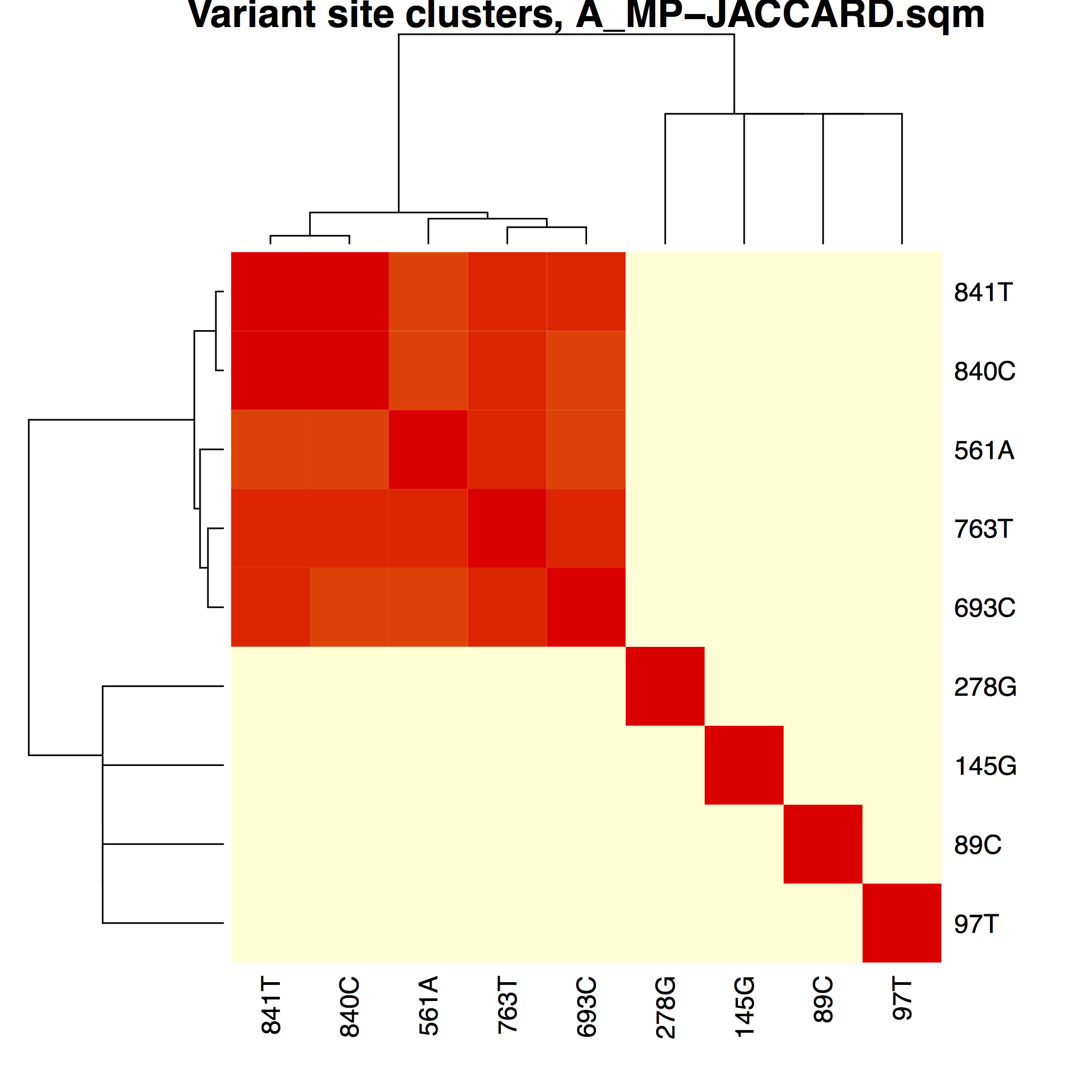
│ ├── A_MP-JACCARD.pdfA_MP variant phasing using modified Jaccard distances
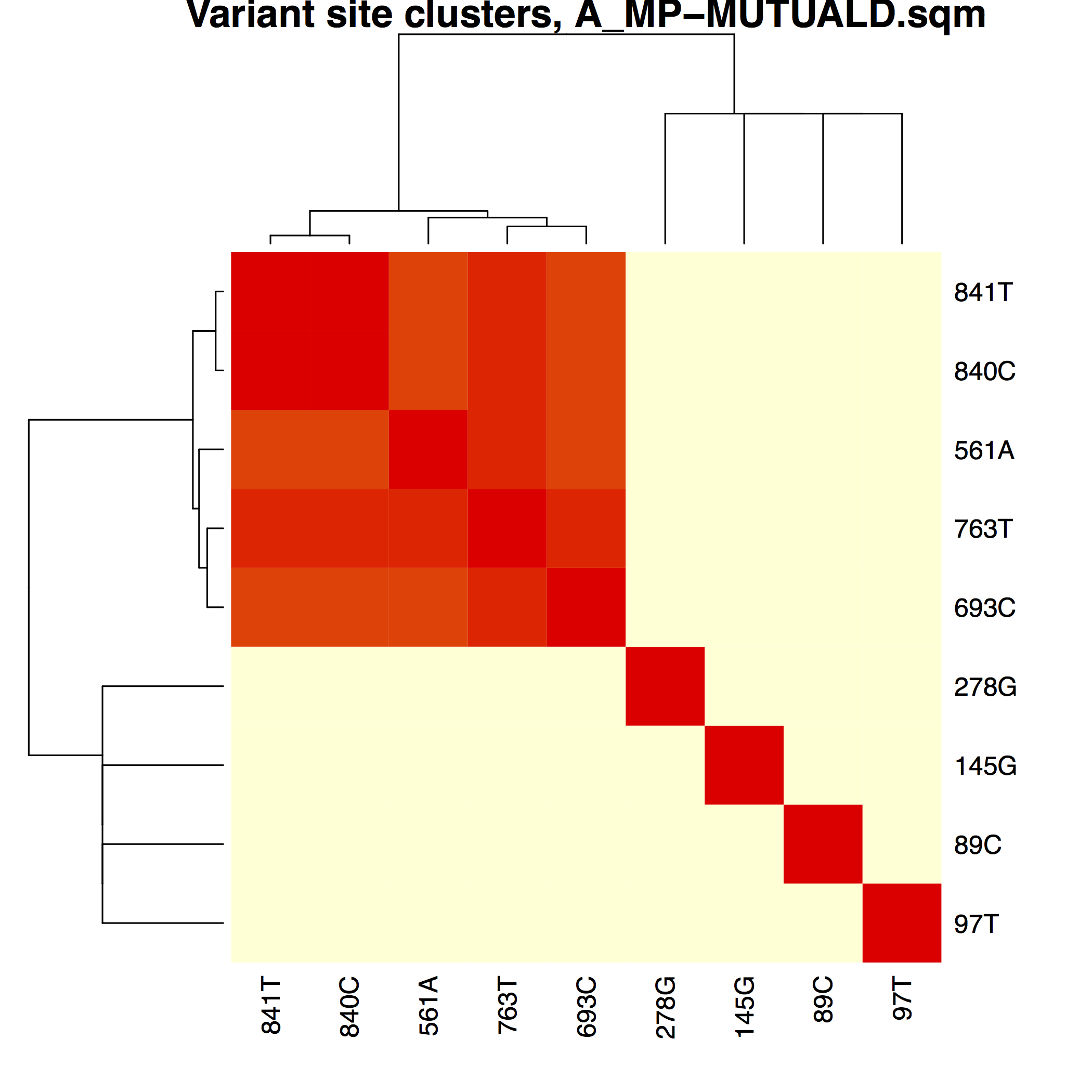
│ ├── A_MP-MUTUALD.pdfA_MP variant phasing using mutual association distances
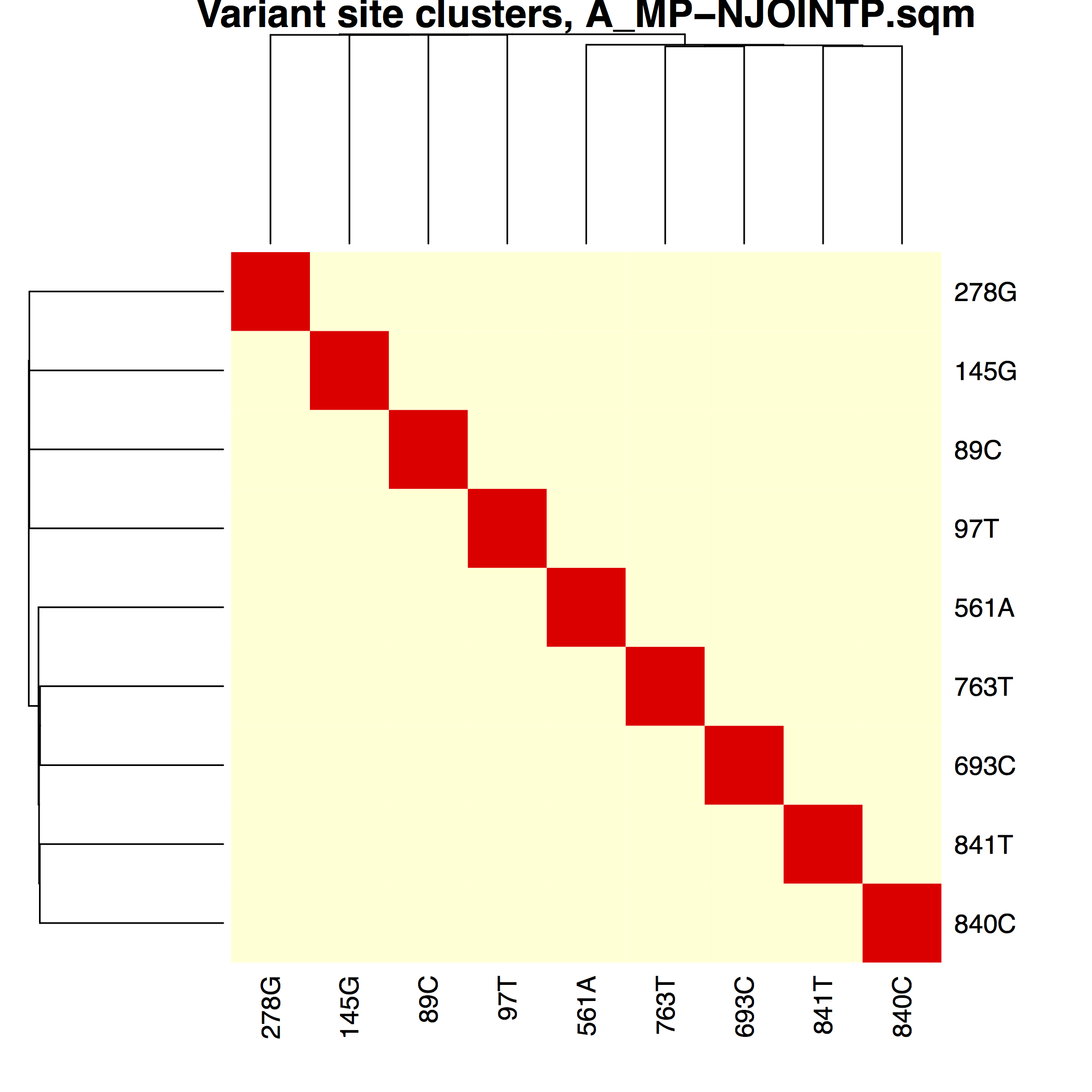
│ ├── A_MP-NJOINTP.pdfA_MP variant phasing using normalized joint probability
distances
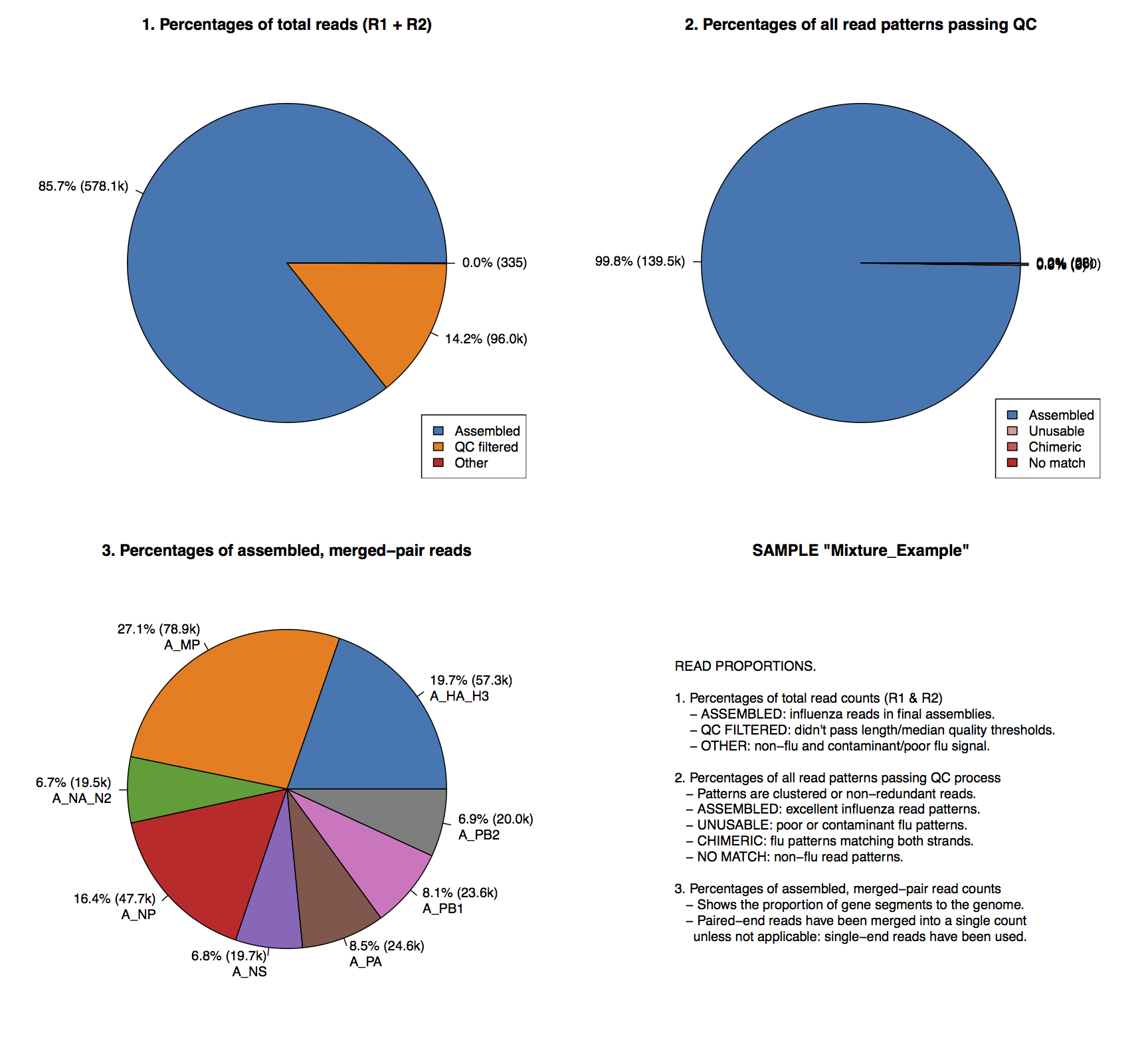
│ └── READ_PERCENTAGES.pdfBreak down for reads assembled.
│
├── intermediate/Intermediate data for each
step
│ ├── 0-ITERATIVE-REFERENCES/
│ │ ├── R0-A_MP.refStarting reference
library sequence for A_MP
│ │ ├── R1-A_MP.refWorking reference for
A_MP after round 1, template for round 2
│ │ └── R2-A_MP.refWorking reference for
A_MP after round 2
│ ├── 1-MATCH_BLAT/
│ │ ├── R1.tar.gz
│ │ ├── R2.tar.gzArchive of BLAT results
for the MATCH step.
│ │ └── R3.tar.gz
│ ├── 2-SORT_BLAT/
│ │ ├── R1.tar.gzClassification/sorting
intermediate files for round 1
│ │ ├── R1.txtSummary statistics of
sorting results for round 1
│ │ ├── R2.tar.gz
│ │ └── R2.txt
│ ├── 3-ALIGN_SAM/
│ │ └── storedCounts.tar.gzStatistic
files used to create rough assembly consensus sequences
│ └── 4-ASSEMBLE_SSWFinal assembly phase
intermediate iterative resuls
│ ├── F1-A_MP.bam
Unsorted BAM file for A_MP assembly, iteration 1
│ ├── F1-A_MP.ref
Reference for final assembly, A_MP, iteration 1
│ ├── F2-A_MP.bam
│ ├── F2-A_MP.ref
│ └── reads.tar.gz
Archive of sorted, unmerged reads by gene segment
│
├── logs/
│ ├── ASSEMBLY_log.txtSSW scores per all
rounds tried in the iterative refinement
│ ├── NR_COUNTS_log.txtRead pattern counts
at various stages
│ ├── QC_log.txtQuality control output
│ ├── READ_log.txtCounts of assembled reads
from BAM files
│ ├── FLU-Mixture_Example.shConfiguration
file corresponding to this IRMA run
│ └── run_info.txtTable of parameters used
by the IRMA run
│
├── matrices/Phasing matrices used to
generate heat maps
│ ├── A_MP-EXPENRD.sqm
│ ├── A_MP-JACCARD.sqm
│ ├── A_MP-MUTUALD.sqm
│ └── A_MP-NJOINTP.sqm
│
├── secondary/
│ ├── R1-A_NA_N1.faTrace A_NA_N1
sorted into secondary status
│ ├── R1-UNRECOGNIZABLE.faRead patterns
that matched flu but had poor signal according to LABEL
│ ├── R2-UNRECOGNIZABLE.fa
│ └── unmatched_read_patterns.tar.gzArchive
of left over read patterns that did not match FLU
│
├── tables/
│ ├── A_MP-pairingStats.txtSummary of paired-end merging statistics, if applicable, A_MP
│ ├── A_MP-coverage.txtSummary coverage statistics for the assembly, A_MP
│ ├── A_MP-coverage.a2m.txtOptional
coverage statistics for plurality consensus globally aligned to profile HMM.
†
│ ├── A_MP-coverage.pad.txtOptional
coverage statistics for padded plurality consensus globally aligned to profile HMM.
†
│ ├── A_MP-allAlleles.txtStatistics for every position & allele in the assembly,
A_MP
│ ├── A_MP-insertions.txtCalled insertion variants for A_MP
│ ├── A_MP-deletions.txtCalled deletion variants for A_MP
│ ├── A_MP-variants.txtCalled single nucleotide variants for A_MP
│ └── READ_COUNTS.txtRead counts for various points in the assembly process
│
├── A_MP.bamSorted BAM file for the final
A_MP assembly (merged if applicable)
├── A_MP.bam.baiBAM file index for A_MP
assembly
├── A_MP.fastaFinal assembled plurality
consensus (no mixed base calls) for A_MP
├── A_MP.a2mOptional plurality consensus
aligned to profile HMM.
†
├── A_MP.vcfCustom variant call file for
called IRMA variants, A_MP
│
├── residual_assembly/Optional residual assembly results †
└── secondary_assembly/Optional secondary assembly results †
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
† optional output
Return to the Influenza Division Bioinformatics Team homepage